Range Operations¶
This section will describe additional GRanges operations provided by gUtils.
## make some example data sets
ref19 <- readRDS(system.file("extdata","refGene.hg19.gr.rds", package="gUtils"))
gr <- GRanges(1, IRanges(c(2,5,10), c(4,9,16)), seqinfo=Seqinfo("1", 20))
gr2 <- c(gr, GRanges(1, IRanges(c(1,9), c(6,14)), seqinfo=Seqinfo("1", 20)))
dt <- data.table(seqnames=1, start=c(2,5,10), end=c(3,8,15))
shift(gr, 2)
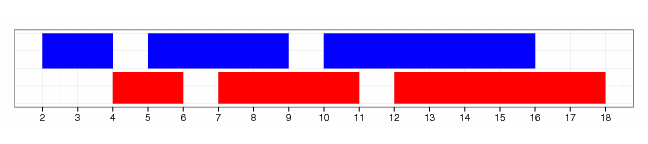
flank(gr, width=2)
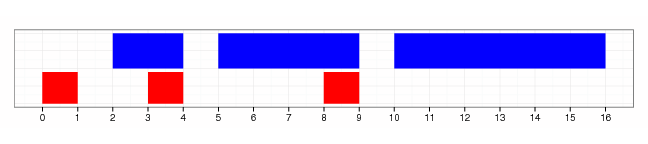
gr.start(gr, width=3)
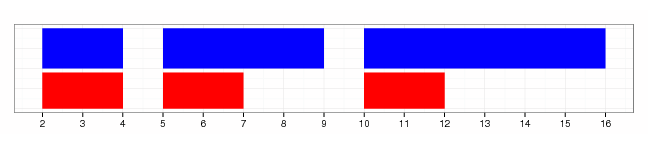
gr.end(gr, width=3)
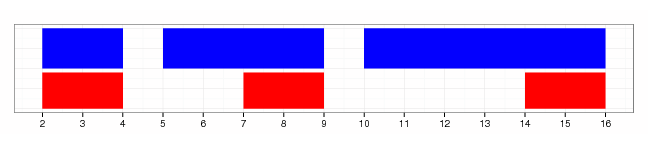
gr.mid(gr) + 2

grbind
## add metadata to one field
mcols(gr)$score = 3
## try to concatenate
c(gr,gr2) ## ERROR
## with grbind
grbind(gr, gr2) ## SUCCESS. Adds NA for missing fields
## GenomicRanges::c does this already for GRangesList
streduce(gr2)

gr.sample(gr2, 2, len=2, replace=TRUE)
## output GRanges
GRanges object with 3 ranges and 1 metadata column:
seqnames ranges strand | query.id
<Rle> <IRanges> <Rle> | <integer>
[1] 1 [ 8, 9] * | 2
[2] 1 [ 5, 6] * | 2
[3] 1 [11, 12] * | 3

gr.rand(w=c(2,5,3), seqinfo(gr))
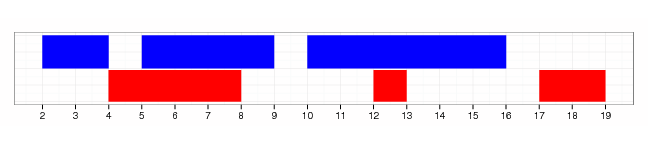
gr.simplify
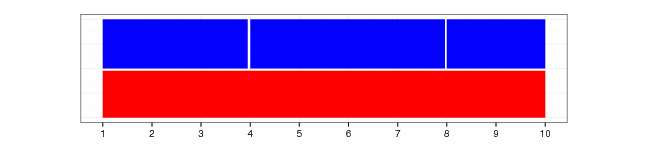
gr.tile(GRanges(1, IRanges(1,9)), w=3) + 1
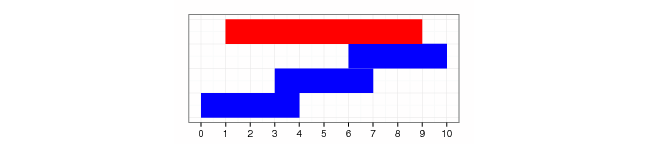
gr.refactor
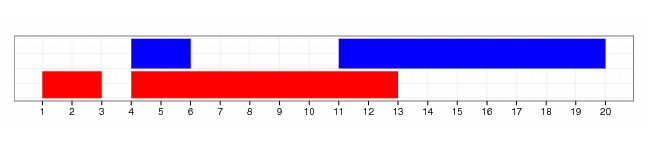
gr.tile.map
gr.round